Псевдогипопаратиреоз симптомы. Что означает диагноз гипопаратиреоз? Симптомы и признаки гипопаратиреоза
Псевдогипопаратиреоз - достаточно редкое заболевание костной системы, суть которого - нарушение обмена кальция и фосфора. У больного обычно наблюдается торможение умственного и физического развития. Болезнь имеет наследственный характер.
Из того, что в названии болезни есть приставка «псевдо», можно легко понять, что она имитирует гипопаратиреоз. Второе название этого недуга - наследственная остеодистрофия Олбрайта, по фамилии врача, который еще в середине прошлого столетия изучил и описал эту патологию.
Типы псевдогипопаратиреоза
Есть два типа псевдогипопаратиреоза - в зависимости от того, поменялся уровень кальция в крови или нет. У первого типа болезни Олбрайта - те же клинические признаки, что и у идиопатического гипопаратиреоза, при этом характерно уменьшение содержания кальция в крови. Чувствительности тканей к паратгормону нет. Второй тип псевдогипопаратиреоза, наоборот, отличается тем, что уровень кальция - в норме. Поэтому этот тип именуют псевдопсевдогипопаратиреозом. Кстати, по данным эндокринологов, одна форма запросто может перейти в другую, к тому же у членов одной семьи могут быть разные типы заболевания.
Причины псевдогипопаратиреоза
Нарушения фосфор-кальциевого метаболизма развиваются из-за сопротивляемости тканей к паратгормону, который продуцируют паращитовидные железы. На основе псевдогипопаратиреоза впервые был подтвержден феномен нарушения чувствительности тканей к гормону, который производят железы внутренней секреции или который вводится извне.
Псевдогипопаратиреоз - патология генетического характера. Вызывает его врожденный синдром - специфический клеточный рецептор - паратгормон-аденилатциклаза, именно из-за него периферические ткани теряют чувствительность к паратгормону.
Кто более всего предрасположен к псевдогипопаратиреозу? В первую очередь - дети больного, другие родственники, поскольку заболевание имеет наследственный аутосомно-доминантный характер. Женщины страдают от болезни Олбрайта чаще. Специалисты объясняют это тем, что у мужчин с псевдогипопаратиреозом - низкая плодовитость, а сама наследственность лишь в единичных случаях переходит от отца к сыну.
Насколько часто встречается это заболевание - статистики нет, в медицинских трудах описано около трехсот случаев.
Симптомы псевдогипопаратиреоза
- Неполадки в органах эндокринной системы при псевдогипопаратиреозе обуславливают и основные признаки: больной обычно коренастый, ниже ростом, чем другие, из-за укороченных нижних конечностей, страдает ожирением, уровень глюкозы в крови - повышен, лицо приобретает лунообразную форму;
- У представительниц слабого пола нарушается менструальный цикл. Нередко эти признаки сопровождаются акромегалией, гинекомастией, различными аутоиммунными процессами, синдромом Иценко-Кушинга и другими недугами. Выше у таких больных и риск возникновения сахарного и несахарного диабета;
- Страдающие болезнью Олбрайта нередко жалуются на проблемы с зубами (у них повреждается эмаль), также больных беспокоят рвота, тонические судороги - как спонтанные, так и возникающие под действием раздражителей, в крови может наблюдаться моча. Есть риск возникновения катаракты. Характерна умственная отсталость. Дети с такой болезнью не справляются со школьной программой, у них снижена память;
- В костно-суставной системе также происходят значительные изменения - проявляется диффузный остеопороз, развиваются кисты (бурые опухоли). Больные псевдогипопаратиреозом чаще других страдают от переломов или деформации костей. У них укорачиваются первая, четвертая и пятая пястные и плюсневые кости, на кистях и стопах наблюдаются признаки рассасывания костей. К тому же, кальций, который освободился из костей, начинает накапливаться в подкожной клетчатке, образуя кальцинаты (такое бывает еще при оссифицирующем миозите), в зоне суставов, полости черепа. Кальцинаты также присутствуют в мышцах, почках, в миокарде и на стенках больших артерий;
- Еще один отличительный признак болезни Олбрайта - пигментация кожи. По телу (как с одной стороны, так и с другой стороны) распространяются коричневые пятна.
Касательно трудоспособности, то она зависит от тяжести процесса и эффективности медикаментозного лечения. Если форма заболевания скрытая и нет явных приступов, то трудоспособность частично сохранена. Но есть ряд ограничений - нельзя, в частности, работать с движущимися механизмами, на транспорте, запрещено и нервно-психическое перенапряжение, чрезмерный физический труд. Если бывают частые судорожные приступы, явно выражена умственная отсталость, значительное нарушение зрения вследствие катаракты, то такие больные уже считаются нетрудоспособными.
Диагностика псевдогипопаратиреоза
Симптомы достаточно специфические и если они начали проявляться, то немедленно надо обращаться к квалифицированному врачу. Именно ранняя и правильная диагностика обеспечивает успешность лечения.
Поскольку болезнь Олбрайта - врожденная, то обычно ее диагностируют больному еще в детстве (дошкольном, младшем школьном возрасте). Выводы врачи-генетики делают на основе клинических симптомов, также применяются лабораторные и инструментальные диагностические методы.
В частности, пациент для уточнения диагноза сдает кровь (у больных псевдогипопаратиреозом уровень кальция низкий, а фосфора и паратгормона - высокий, высокая и активность щелочной фосфатазы в крови), мочу (у больного этот анализ покажет уменьшение выделенных фосфора и кальция). Делаются и специальные пробы, которые показывают, какая у пациента чувствительность тканей почечных канальцев к паратгормону. Иногда возникает необходимость в оценке уровня оксипролина.
Инструментальные методы предполагают рентгенодиагностику (она наиболее информативна при выявлении специфических изменений в костной и мышечной тканях).
Лечение псевдогипопаратиреоза
Эту редкую наследственную патологию лечат препаратами кальция. Дозу подбирает врач, исходя из содержания кальция, нужного для поддержания гомеостаза. А чтобы кальций без проблем усваивался, то в терапевтическую схему еще и включают препараты витамина Д. Кроме того, врач для больного псевдогипопаратиреозом разработает диету, в которой будет ограничено количество продуктов, содержащих фтор. Эта диета поможет привести в норму концентрацию в крови кальция и убрать признаки вторичного гиперпаратиреоза.
Если болезнь Олбрайта сопровождается другими эндокринными нарушениями, то может возникнуть потребность в гормональной заместительной терапии. Судороги снимают с помощью раствора хлористого или глюконата кальция, который вводят внутривенно. В случае появления катаракты ее лечением занимается окулист, а психолог помогает снять когнитивные нарушения.
При продуманной схеме терапии прогнозы очень благоприятны. Но все переболевшие псевдогипопаратиреозом регулярно должны консультироваться у врачей-генетиков, чтобы предупредить заболевание у потомков.
Профилактика псевдогипопаратиреоза
Учитывая, что болезнь наследственная, то единственный способ профилактики для тех, у кого в роду был псевдогипопаратиреоз - это регулярное посещение медицинских генетиков для консультаций.
(болезнь Олбрайта) - наследственная остеодистрофия, обусловленная резистентностью периферических тканей к паратгормону, что сопровождается расстройством кальциево-фосфорного обмена, задержкой физического и умственного развития. Псевдогипопаратиреоз протекает с явлениями диффузного остеопороза, тоническими судорогами, переломами и деформацией костей, отложением кальция в мышцах и сосудах, образованием камней в мочевыводящих путях, задержкой роста и отставанием умственного развития. С целью диагностики псевдогипопаратиреоза проводятся определение уровня кальция, щелочной фосфатазы, паратгормона в крови; экскреции фосфора и кальция с мочой; функциональная проба с введением паратгормона; рентгенодиагностика. Лечение псевдогипопаратиреоза предполагает медикаментозную коррекцию гипокальциемии.
МКБ-10
E20.1

Общие сведения
Псевдогипопаратиреоз является редкой наследственной патологией; в эндокринологии известно всего около 300 случаев данного заболевания. Псевдогипопаратиреоз по своим проявлениям напоминает гипопаратиреоз . Однако, если в основе гипопаратиреоза лежит первичный дефицит паратиреоидного гормона, обусловленный снижением функциональной активности паращитовидных желез, то при болезни Олбрайта псевдогипопаратиреоидный синдром вызывается нарушением чувствительности тканей-мишеней к действию паратгормона при его достаточном уровне секреции.
Впервые псевдогипопаратиреоз был описан в 1942г. Ф.Олбрайтом и по автору получил название «болезни Олбрайта» или «наследственной остеодистрофии Олбрайта». Первый вариант заболевания характеризуется аномалиями развития скелета, резистентностью периферических тканей к паратгормону, гипокальциемией и клинической картиной, сходной с идиопатическим гипопаратиреозом. Второй вариант болезни Олбрайта протекает по нормокальциемическому типу и получил в литературе название псевдопсевдогипопаратиреоза. Описаны случаи развития в одной семье как нормо-, так и гипокальциемических вариантов заболевания, а также переход одной формы в другую.
Причины псевдогипопаратиреоза
При любых вариантах псевдогипопаратиреоза заболевание носит наследственный характер с аутосомно-доминантным типом наследования, сцепленным с X-хромосомой. Изучение родословных показывает, что число женщин с псевдогипопаратиреозом в 2 раза превышает количество больных мужчин; кроме того, болезнь Олбрайта не передается от отца к сыновьям.
Псевдогипопаратиреоз обусловлен генетической резистентностью скелета и почек к действию паратиреоидного гормона. Это связано с дефектом специфических рецепторов плазматических мембран клеток-мишеней и недостаточностью ферментов аденилатциклазы, протеинкиназы, циклического 3, 5- аденозинмонофосфата (АМФ) в дистальных канальцах нефрона, что сопровождается реабсорбцией фосфора. Снижение выделения фосфора с мочой приводит к гиперфосфатемии и вторичной гипокальциемии.
Поскольку при псевдогипопаратиреозе паращитовидные железы остаются интактными, гипокальциемия может вызывать стимуляцию секреции паратгормона и развитие вторичного гиперпаратиреоза . При псевдогипопаратиреозе обычно имеет место компенсаторная гиперплазия паращитовидных желез (развитие аденом не типично), изменения со стороны костной ткани (остеопороз , кисты), отложение кальцинатов в скелетных мышцах, подкожной клетчатке, а также в почках, стенках артерий, миокарде, конъюнктиве и роговицы глаза. Псевдогипопаратиреоз часто сочетается с артериальной гипертензией , сахарным диабетом , артериитом , полиартритом .
Симптомы псевдогипопаратиреоза
Клинические проявления псевдогипопаратиреоза аналогичны признакам идиопатического гипопаратиреоза. Эндокринные симптомы при псевдогипопаратиреозе включают низкорослость, ожирение и гипергликемию, лунообразное лицо, олигоменорею . Нередко болезнь Олбрайта сочетается с другими эндокринными нарушениями - акромегалией , синдромом Кушинга , гинекомастией , гипотиреозом или гипертиреозом .
Костно-суставные признаки псевдогипопаратиреоза включают брахидактилию с заметным укорочением 1, 4 и 5 пястных и плюсневых костей; экзостозы, дисхондроплазию, изменения в эпифизарных концах костей, резорбцию костей пальцев рук. Характерны задержка прорезывания зубов , гипоплазия зубной эмали , подкожные оссификаты.
Неврологическая симптоматика псевдогипопаратиреоза представлена приступами тонических судорог, которые могут возникать спонтанно или под воздействием любых раздражителей. Для больных с псевдогипопаратиреозом типично отставание в умственном развитии. Среди прочих симптомов псевдогипопаратиреоза отмечается мочекаменная болезнь , гематурия, рвота, развитие лентикулярной катаракты . При псевдопсевдогипопаратиреозе отсутствуют гипокальциемия, гиперфосфатемия, остеомаляция и судорожный синдром.
Диагностика псевдогипопаратиреоза
В типичных случаях псевдогипопаратиреоз диагностируется у детей в возрасте 5-10 лет с учетом характерной симптоматики, лабораторных и рентгенологических данных. У больных с псевдогипопаратиреозом определяется гипокальциемия, гиперфосфатемия, нормальная или повышенная активность щелочной фосфатазы в крови, увеличенное содержание паратгормона в сыворотке крови; уменьшение экскреции кальция и фосфора с мочой.
Нечувствительность почечных канальцев к паратиреоидному гормону подтверждается с помощью теста, основанного на определении количества выводимых с мочой фосфатов и циклического аденозинмонофосфата. При псевдогипопаратиреозе в ответ на внутривенное введение паратгормона достоверное повышение содержания фосфатов и цАМФ в моче отсутствует.
При псевдогипопаратиреозе необходима коррекция гипокальциемии, в связи с чем проводится терапия препаратами кальция в дозировках, позволяющих поддерживать нормальную концентрацию кальция в крови. Показан прием витамина D и его активных форм под контролем концентрации кальция в сыворотке крови. Лечение паратиреоидным гормоном не эффективно. Для нормализации концентрации кальция и устранения симптомов вторичного гиперпаратиреоза необходимо соблюдение диеты с ограничением употребления фосфора.
При функциональной недостаточности других желез проводится заместительная терапия соответствующими гормонами. При возникновении судорог показано внутривенное введение растворов кальция.
Своевременная квалификация и рациональная терапия псевдогипопаратиреоза позволяет говорить о положительных прогнозах на жизнь и возможности контроля за течением заболевания. Учитывая генетический характер псевдогипопаратиреоза, целесообразно проведение медико-генетического консультирования для оценки риска появления болезни Олбрайта у потомства.
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., И.В. Шулякова, невролог, к. м. н.,
обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Минздрава РФ, г. Москва
Ключевые слова:
дети, псевдогипопаратиреоз, наследственная остеодистрофия Олбрайта, ожирение, гипокальциемия, диагностика, резистентность к паратиреоидному гормону.
Keywords:
children, pseudohypoparathyroidism, Albright hereditary osteodystrophy, obesity, hypocalcemia, diagnostics, parathyroid hormone resistance.
Псевдогипопаратиреоз (греч. pseudes – ложный + гипопаратиреоз; синоним: наследственная остеодистрофия Олбрайта, синдром «яванской курицы») – редкое наследственное заболевание костной системы, имитирующее гипопаратиреоз и характеризующееся нарушением обмена кальция и фосфора; часто сопровождается задержкой умственного и физического развития. Заболевание описано впервые американским врачом-эндокринологом Albright F. в 1942 году . Распространенность заболевания составляет 7,9 на 1 млн человек .
ГЕНЕТИЧЕСКИЕ ДАННЫЕ
Псевдогипопаратиреоз (ПГП) – генетически гетерогенное заболевание. Данные о типе наследственной передачи противоречивы: как X-сцепленный доминантный , так и аутосомно-доминантный, аутосомно-рецессивный типы . В большинстве случаев развитие наследственной остеодистрофии Олбрайта связано с мутациями в расположенном на хромосоме 20 локусе 20q13 гена GNAS1 (Patten et al., 1990), кодирующего белок Gs-альфа, связанного с рецептором паратиреоидного гормона (ПТГ) . Подобный фенотип выявлен и у больных с интерстициальной делецией длинного плеча хромосомы 2 локуса 2q37 .
ПАТОГЕНЕЗ
В основе патогенеза псевдогипопаратиреоза лежит генетически обусловленная резистентность почек и скелета к действию парат-гормона в результате дефекта комплекса «специфический циторецептор – паратгормон – аденилатциклаза», что нарушает процесс образования в почках циклического 3"-, 5"-аденозинмонофосфата (цАМФ), являющегося внутриклеточным посредником действия паратгормона на метаболические процессы. Псевдогипопаратиреоз является генетически гетерогенным заболеванием. У части больных дефектен сам циторецептор, связывающий паратгормон (тип 1А псевдогипопаратиреоза), у других отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном бислое клеточной мембраны и функционально связывающего рецептор с аденилатциклазой (тип 1B псевдогипопаратиреоза). У некоторых больных наблюдается ферментативная недостаточность самой аденилатциклазы (псевдогипопаратиреоз 2-го типа). Дефицит цАМФ, получающийся вследствие этих дефектов, ведет к нарушению синтеза специфических белков, определяющих биологический эффект паратгормона. Таким образом, теряется чувствительность органов-мишеней к паратгормону .
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
В настоящее время выделяют 4 клинические формы патологии: типы 1А, 1В, 1С и 2. Знание их клинико-биохимических особенностей и данных генетических исследований позволяет провести дифференциальную диагностику в рамках самой нозологической формы.
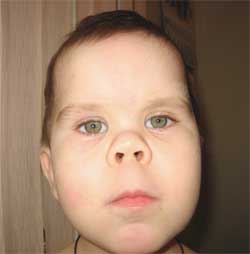
Общими признаками, позволяющими заподозрить заболевание, являются диспропорциональность физического развития, низкий рост (до карликовости) за счет укорочения нижних конечностей (фото 1), брахидактилия (фото 2), круглое «лунообразное» лицо (фото 3). Иногда наблюдаются экзостозы и аплазия зубов.
Фото 1.
Внешний вид ребенка с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост за счет укорочения нижних конечностей)
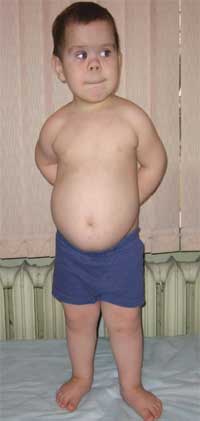
Фото 2.
Особенности костной системы у больного
с остеодистрофией Олбрайта
(брахидактилия – укорочение пальцев)
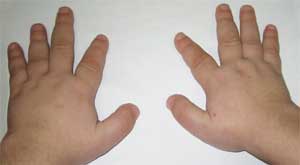
Фото 3.
Особенности фенотипа ребенка
с остеодистрофией Олбрайта
(круглое «лунообразное» лицо)
Патогномоничным признаком считается резкое укорочение I, III и V пястных и плюсневых костей (особенно III и IV), вследствие чего II пальцы на кистях и стопах оказываются длиннее остальных, а при сжатии кисти в кулак отсутствуют выпуклости в области IV и V пястно-фаланговых суставов – так называемый брахиметафалангизм. Выявляются также короткие широкие фаланги, утолщение свода черепа и деминерализация костей (остеопороз), ожирение .
Умственная отсталость (чаще умеренной степени выраженности) обнаруживается примерно у 20% больных. По данным некоторых авторов , олигофрения встречается в 70% случаев при наличии гипокальциемии и в 30% случае при нормокальциемии. Психические процессы у больных замедлены. В неврологическом статусе нередко отмечаются моторная неловкость, невротические реакции: страхи, тревога, беспокойство, плохой сон, повышение рефлексов, судороги, носящие тетанический характер и обусловленные гипокальциемией, иногда судорожные пароксизмы. Описаны также миопатические симптомы: мышечная утомляемость, мышечная слабость. Часто наблюдаются экстрапирамидные нарушения: хореиформные гиперкинезы, атетоз, лицевой гемиспазм, паркинсонизм, в отдельных случаях имеют место эпилептические пароксизмы, мозжечковые симптомы: атаксия, нарушение координации.
Нередко определяются кальцификация мягких тканей, подкожные кальцификаты (грудь, живот, пяточные сухожилия), при гистологическом исследовании которых – osteoma cutis (Izraeli et al., 1992), мозге (базальные ганглии). Важно отметить, что кальцификаты могут быть уже при рождении. Вследствие гипокальциемии обычно развивается катаракта и возникают дефекты эмали зубов.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1А
имеет аутосомно-доминантный тип наследования. Ген псевдогипопаратиреоза типа 1А – GNAS1 – локализован на длинном плече хромосомы 20, в локусе 20q13.2. Развитие заболевания связано с дефицитом гуанин-нуклеотид-связывающего белка (Gs-белок). При этом ПТГ, связываясь с рецепторами тканей-мишеней, не способен активизировать циклический аденозинмонофосфат (цАМФ) и вызвать тканевой ответ. Вероятно, подобный механизм лежит в основе развития нечувствительности тканей других органов и эндокринных желез (гипофункция щитовидной железы, гонад, гипофиза, сахарный диабет, а также сниженный ответ печени на введение глюкагона), наблюдаемой при псевдогипопаратиреозе типа 1А. При данном типе патологии не наблюдается характерной для нормы повышенной экскреции цАМФ с мочой в ответ на экзогенное введение ПТГ. Заболевание диагностируется чаще в возрасте 5–10 лет. У больных наблюдаются низкий рост, короткая шея, круглое лицо, укорочение метакарпальных и метатарзальных костей (чаще укорочение IV и реже II пальцев) – так называемый брахи-метафалангизм. Отмечаются кальцификация мягких тканей, подкожные кальцификаты, которые могут выявляться уже при рождении; нередко наблюдается одновременное вовлечение других эндокринных желез: щитовидной железы (гипофункция), гонад, поджелудочной железы (сахарный диабет). Вследствие гипокальциемии нередко развиваются катаракта и дефект эмали зубов. В качестве дифференциально-диагностического теста отличия ПГП типа 1А от гипопаратиреоза: отсутствие клинического эффекта от парентерального введения ПТГ в виде подъема уровня кальция в крови и увеличения почечной экскреции фосфора с мочой (фосфатурический эффект).
При биохимическом исследовании выявляются гипокальциемия, гиперфосфатемия, увеличение уровня паратиреоидного гормона в крови, гипофосфатурия. Уровень Gs-белка в крови снижен. При рентгенологическом исследовании костной системы обнаруживаются укорочение метакарпальных и метатарзальных костей, генерализованная деминерализация, утолщение костей свода черепа.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1B
имеет аутосомно-доминантный тип наследования, однако не исключен доминантный, сцепленный с Х-хромосомой тип наследования. Необходимо иметь в виду наблюдающуюся иногда неполную пенетрантность гена болезни и возможность скрытого носительства патологии. Поэтому рекомендуется клиническое (выявление субклинического течения болезни) и биохимическое обследование (определение уровней кальция, фосфора, ПТГ крови) предполагаемых носителей заболевания. ПГП типа 1В обусловлен дефицитом тканевых рецепторов к паратиреоидному гормону в органах-мишенях и ограниченной резистентностью к паратгормону. Клиническая картина сходна с клиникой типа 1А, но отсутствует поражение других эндокринных желез, реже встречается остеодистрофия.
У больных отсутствует реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой, однако, в отличие от типа 1А, уровень Gs-белка в крови нормален. Женщины поражаются чаще мужчин, однако тяжесть заболевания может быть одинаковой как у мужчин, так и у женщин.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1С
некоторые авторы отождествляют с псевдо-псевдогипопаратиреозом (ППГП), описанным Albright F. в 1952 году. Характеризуется свойственной ПГП клинической картиной, однако уровни кальция, фосфора в крови и моче остаются в пределах нормы. Показатели ПТГ и Gs-белка в крови также сохраняются на нормальном уровне. У некоторых больных с ПГП типа 1С обнаруживаются делеции de novo на хромосоме 2 . Не исключено, что этот вариант болезни является подтипом ПГП типа 1А.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 2
клинически сходен с другими типами заболевания, однако имеет аутосомно-рецессивный тип наследования. Не исключено существование и аутосомно-доминантных форм патологии. Патогенез развития связан с внутриклеточной резистентностью к цАМФ. ПТГ при этом связывается с рецепторами и вызывает нормальную ответную реакцию клеток на ПТГ в виде увеличения экскреции цАМФ. Внутриклеточная нечувствительность к цАМФ, однако, не позволяет осуществиться полной реализации действия ПТГ. При этом сохраняется нормальная реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой. Высказывается мнение, что ПГП типа 2 может быть связан с дефицитом витамина D .
Таким образом, выделенные типы ПГП клинически характеризуются пониженной чувствительностью органов-мишеней к ПТГ, однако различаются патогенетическими механизмами формирования нечувствительности тканей.
ДИАГНОСТИКА
Лабораторным дифференциально-диагностическим тестом может служить характер почечной экскреции цАМФ в ответ на введение ПТГ: повышенная экскреция цАМФ отмечается при типе 2 и ее отсутствие – при типе 1. Диагноз подтверждается обнаружением сниженного уровня гуанин-нуклеотидсвязывающего белка (Gs-белок) в крови (в среднем в 1,5–2 раза) по сравнению с нормой. Гипокальциемия, как правило, сочетается с гиперфосфатемией и гипофосфатурией. Уровень ПТГ повышен; при 1C-типе уровень ПТГ в норме, что дало основание для названия «псевдогипопаратиреоз». При рентгенологическом исследовании костной системы обнаруживаются укорочение пястных и плюсневых костей, нередко генерализованная деминерализация (остеопороз), утолщение костей свода черепа. В дерматоглифическом рисунке отмечается смещение осевого ладонного трирадиуса.
Критерии диагноза:
- низкий рост;
- круглое лицо;
- задержка нервно-психического развития;
- скелетные аномалии;
- низкое содержание кальция в сыворотке крови;
- высокий уровень паратиреоидного гормона в крови;
- снижение экскреции с мочой фосфатов и цАМФ.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечение при гипокальциемии заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови. Большое значение имеет терапия витамином D. В настоящее время применяют активные метаболиты витамина D – оксидевит, 1-альфа-Д3, кальцитрин и др. в дозе 1–2 мкг/сутки с положительным результатом (увеличение содержание кальция в крови, уменьшение проявлений судорожного синдрома). Эффективен также тахистин (0,5–1,5 мг/сутки). Данный препарат увеличивает всасывание кальция в кишечнике и тем самым способствует повышению уровня кальция в крови. Противосудорожная терапия используется как дополнительное лечение. На интеллектуальное развитие лечение не оказывает заметного действия, но наряду с уменьшением симптомов судорожного синдрома наблюдается регресс неврологических проявлений (подкорковых нарушений, хореиформных гиперкинезов, атетоза и др.). Во избежание передозировки препаратов витамина D необходим контроль концентрации кальция в крови каждые 3–7 дней в течение первых 2 недель лечения и каждый месяц в течение последующих 2–3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2–3 месяца. Диета с ограничением фосфора помогает нормализовать как концентрацию фосфора, так и содержание кальция в крови и устранить симптомы вторичного гиперпаратиреоза. При недостаточности других желез внутренней секреции проводят заместительную терапию соответствующими гормонами.
Лечение паратгормоном неэффективно. Для купирования судорожных приступов внутривенно вводят 10%-ный раствор кальция хлорида или кальция глюконата; внутрь – 5–10%-ный раствор кальция хлорида по 1 столовой ложке 3–4 раза в день: кальция глюконат, кальция лактат – до 10 г в день.
ПРОГНОЗ для жизни определяется выраженностью судорожного синдрома.
ПРОФИЛАКТИКА болезни основывается на данных медико-генетического консультирования.
МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в гене GNAS1 на хромосоме 20. Разрабатываются способы пренатальной диагностики заболевания в целом и отдельных его типов.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ Мальчик Г., 14,5 лет (фото 4), поступил в Научно-исследовательский клинический институт педиатрии с диагнозом: дегенеративное заболевание нервной системы? врожденная наружная гидроцефалия; симптоматическая эпилепсия; наследственный синдром? болезнь накопления? метаболическая энцефалопатия; субклинический гипотиреоз; низкорослость смешанного генеза; когнитивные нарушения.
Жалобы при поступлении на интенсивные приступообразные головные боли, локализующиеся в лобной области и сопровождающиеся рвотой, которая приносит облегчение, снижение памяти и успеваемости в школе, судорожные приступы, во время которых происходит подергивания в правой руке.
Фото 4.
Ребенок Г., 14,5 лет, с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост, укорочение конечностей, брахидактилия)
Анамнез семейный: родители – армяне по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии – не отмечалось. Сибс, сестра 17 лет – со слов – здорова.
Анамнез жизни и заболевания:
мальчик от 2-й беременности, протекавшей без особенностей, роды вторые, в срок, физиологические, масса при рождении – 3100 г, длина – 51 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. Ухудшение состояния на 3-и сутки – судороги неонатальные, купированы в роддоме. Ранний постнатальный период – без особенностей. Отмечалась незначительная темповая задержка моторного развития на первом году жизни, самостоятельная ходьба с 1 года 3 мес. В связи с чем наблюдался неврологом с диагнозом: органическое поражение ЦНС; врожденная гидроцефалия; неонатальные судороги; фебрильные судороги в анамнезе.
Получал диакарб, финлепсин. Дебют приступов с 1 года 11 мес. – асимметричные, тонические в виде напряжения правой руки и ноги, с заведением глаз, до 2 мин., без потери сознания, частые до 10 эпизодов в сутки. Получал депакин нерегулярно. На фоне самостоятельной отмены – однократный тонический статус. В 2 года проведена по месту жительства КТ головного мозга, где выявлены единичные очаги демиелинизации в затылочных долях.
Консультирован нейрохирургом, рекомендовано консервативное лечение. С 3 лет отмечается задержка психоречевого развития, рекомендовано наблюдение психиатра.
С 4–5 лет родители стали отмечать деформацию и укорочение пальцев стоп и кистей, в особенности II–IV пальцев симметрично на руках и ногах, снижение ростовых показателей. В 8 лет заключение логопеда – общее нарушение речи 2–3-го уровня, рекомендовано обучение в специализированной школе. В этом же возрасте осмотр генетиком по месту жительства, заключение: наследственная болезнь обмена? рекомендовано исследование аминокислот крови, изменений не было выявлено; окончательное заключение: данных за наследственное заболевание обмена не выявлено; гипохондроплазия; рекомендовано лечение невролога и эндокринолога.
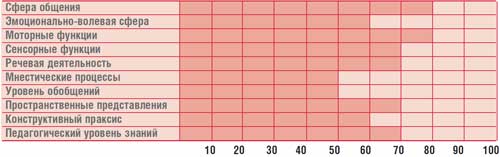
Таблица.
Профиль психического развития ребенка Г., 14,5 лет (IQ = 68)
В возрасте 8 лет консультирован эндокринологом по поводу задержки роста и развития. При рентгенологическом исследовании кистей рук отмечены особенности: средние, основные фаланги и пястные кости укорочены, утолщены; диагноз рентгенолога – ахондроплазия.
Неоднократно обследуется по месту жительства в неврологическом стационаре. В 12 лет появились судорожные приступы без потери сознания с подергиванием правой руки, носящие серийный характер, назначена противосудорожная терапия (депакин), частота приступов значительно сократилась. В 13 лет проведена МРТ головного мозга с контрастированием – симметричные изменения в основании височных долей на уровне ядер в виде повышения МР-сигнала, что характерно для токсических (марганец) или метаболических (медь, железо) энцефалопатий.
Вновь осмотрен в возрасте 13 лет 3 мес. эндокринологом, при исследовании тиреоидного профиля выявлено повышение тиреотропного гормона (ТТГ), диагностирован субклинический гипотиреоз, назначен L-тироксин.
При анализе амбулаторной карты ребенка и документации по месту жительства исследование кальция и фосфора проводилось однократно, в возрасте 1,5 лет, отмечалась гипокальциемия, но по данному поводу дообследование не проводилось. Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Москву, в Научно-исследовательский клинический институт педиатрии, с целью уточнения диагноза.
Данные объективного исследования:
Рост – 143 см, масса – 43 кг.
Физическое развитие очень низкое, гармоничное, телосложение диспропорциональное за счет укорочения конечностей. Sds роста соответствует –2,8 отклонениям от нормы (норма –2+2).
Особенности фенотипа: круглое лицо, короткая шея, антимонголоидный разрез глазных щелей, широкое переносье, высокий лоб, брахидактилия, укорочение IV и V пястных и плюсневых костей (фото 5). По внутренним органам – без особенностей. Половое развитие – Tanner III–IV стадия (что соответствует возрасту).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови: общий кальций – 1,39 (норма 2,02–2,6 ммоль/л), кальций ионизированный – 0,61 (норма 1,13–1,32 ммоль/л), фосфор неорганический – 3,66 (норма 0,86–1,56 ммоль/л), остальные показатели в пределах нормы.
Биохимический анализ мочи: почечная экскреция фосфатов снижена – 11,5 ммоль/л (норма 19–32 ммоль/л).
Тиреоидный профиль: ТТГ – 11,75 (норма 0,4–4,0 мкМЕ/мл), свободный Т4 – 0,49 (норма 1,0–1,8 нг/дл).
Паратиреоидный гормон – 499 (норма 12– 65 пг/мл), СТГ – 7 нг/мл (норма 7–10 нг/мл), соматомедин-С – 250 нг/мл (норма 88–360 нг/мл).
УЗИ внутренних органов – без особенностей.
ЭКГ – миграция суправентрикулярного водителя ритма на фоне регулярной ЧСС 71– 80 уд/мин. Неполная блокада правой ножки пучка Гиса. Нарушение процесса реполяризации в миокарде задней стенки левого желудочка (снижение з.Т III, аVF).
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника 1-й степени, выраженный остеопороз.
R-графия кистей рук с захватом предплечий – укорочение и расширение концевых и средних фаланг. Костный возраст – 13,5– 14 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – МР-картина множественных субкортикальных очагов повышенного МР-сигнала в лобных долях, наружная компенсированная гидроцефалия с атрофией вещества головного мозга.
МСКТ головного мозга – симметричные участки обызвествления лентиформных ядер. Диффузные гиперденсивные участки в таламусах, хвостатых ядрах с участком обызвествления справа. Множественные точечные обызвествления покровных мягких тканей черепа.
Аудиограмма – без патологии.
ДНК-диагностика в гене GNAS1 – в работе.
Консультации специалистов:
Эндокринолог – наследственная остеодис-трофия Олбрайта типа 1А (псевдогипопара-тиреоз). Первичный гипотиреоз, неполная медикаментозная компенсация.
Окулист – катаракта полная вторичная. Рекомендовано оперативное лечение.
Психолог – когнитивные нарушения (психологический профиль ребенка представлен в табл.).
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований (гипокальциемия, гиперфосфатемия, гипофосфатурия, повышение паратиреоидного гормона крови), кальцинаты в веществе головного мозга, наличие катаракты, гипотиреоза), поставлен диагноз: наследственная остеодистрофия Олбрайта 1А-типа (псевдогипопаратиреоз). Рекомендовано проведение ДНК-диагностики - поиск мутаций в гене GNAS1.
Лечение: ребенку рекомендован прием эутирокса в дозе 100 мкг/сутки; активный метаболит витамина Д - альфа-Д3 («Тева») в дозе 2 мкг/сутки; кальций («Сандоз») 2000 мг/сутки; постоянный прием противосудорожной терапии - финлепсин 800 мг/сутки под наблюдением невролога-эпилептолога; занятия с логопедом-дефектологом и психологом; энерготропная терапия (Элькар и коэнзим Q10 в возрастных дозах). Контроль показателей фосфорно-кальциевого обмена, уровня паратгормона.
Таким образом,
представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, важность своевременного исследования простых биохимических параметров (при эпилепсии обязателен неоднократный скрининг показателей фосфорно-кальциевого обмена), исходы поздней диагностики генетически детерминированного заболевания, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждение повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии. Список литературы находится в редакции.
Псевдогипопаратиреоз впервые описан в 1942 г. (синоним - наследственная остеодистрофия).
Развитие заболевания связано с рефрактерностъю тканей (почек и костей) к эндогенному и экзогенному паратгормону при повышенной или нормальной его секреции и гиперплазии паращитовидных желез.
Установлено, что тканевая нечувствительность к паратгормону зависит от снижения активности так называемогогуанин-нуклеотидсвязывающего регуляторного белка, обеспечивающего взаимодействие между рецептором и мембранной аденилат-циклазой и участвующего в активации и реализации функции этого фермента.
При этом нарушается синтез цАМФ.
Выделен псевдогипопаратиреоз I типа, при котором активность регуляторного белка снижена на 40-50%. У таких больных нарушение гормональной чувствительности не ограничивается только паратгормоном, но распространяется и на другие гормоны, зависимые от аденилатциклазной системы, в частности, могут наблюдаться нечувствительность щитовидной железы к тиреотропному гормону с повышением реакции тиреотропного гормона на тиреотропный рилизинг-гормон; резистентность половых желез к лютеинизирующему (ЛГ) и фолликулостимулирующему (ФСГ) гормонам с повышением реакции этих гормонов на ЛГ-рилизинг-гормон при отсутствии клинических признаков первичного гипотиреоза и гипогонадизма. При псевдопшопаратиреозе, по-видимому, часть иммунореактнвного паратгормона не обладает биологической активностью. Данных об образовании антител к паратгормону нет.
Патогенез.
В патогенезе псевдогипопаратиреоза I типа определенную роль играет дефицит эндогенного холекальциферола в связи с нарушением чувствительности к паратгормону и дефицитом цАМФ.
При введении дибутирил-3,5-цАМФ повышается содержание в крови холекальциферола, а в результате лечения препаратами витамина D3 увеличивается чувствительность тканей к паратгормону, подоерживаетоягормокалыщемия, устраняется тетания и усиливается коррекция костных нарушений.
При псевдогипопаратиреозе II типа рецепторная чувствительность к паратгормону ее нарушена.
Активность GN-белка нормальна, паратгормон может стимулировать мембранную аденилатцик-лазу, но предполагается нарушение способности транспортных систем кальция и фосфора реагировать на нормально образующийся цАМФ.
Высказано мнение о существовании при псевдогипопаратиреозе II типа аутоантител к плазматическим мембранам клеток почечных канальцев, блокирующих индуцируемую паратгормоиом фосфатурию, т. е. предполагается аутоиммунный генез псевдоги-попаратиреоза II типа. При этой форме заболевания нарушение гормональной чувствительности ограничено тканями, реагирующими на паратгормон. Других нарушений в этом плане не наблюдается.
При псевдогипопаратиреозе возможно возникновение различных сочетаний изменений с разной степенью их выраженности, что определяется генетически.
У родственников больных псевдогшюпаратиреозом нередко имеются отклонения от нормы общего развития и дефекты скелета без нарушения секреции паратгормона, без патологии кальциево-фосфорного обмена и судорог.
Это так называемый псевдопсевдогипопаратиреоз - метаболически нормальный вариант псевдогипопаратиреоза.
Из-за редкости патологии тип наследования этого заболевания точно не установлен.
Соотношение его частоты у женщин и мужчин - 2:1. Предполагают связанное с Х-хромосомой доминантное наследование 2-х взаимосвязанных нарушений - псевдо- и псевдопсевдогипо-паратиреоза, однако есть случаи прямой передачи псевдогипопаратиреоза от отца к сыну, что свидетельствует о возможности ау-тосомного наследования.
Лечение.
Следует выделить его особенности в период острого приступа тетании и подчеркнуть необходимость поддерживающей систематической терапии в межприступном периоде.
Для лечения гипопаратиреозного криза внутривенно вводят 10%-ный раствор хлорида или глюконата кальция. Доза определяется тяжестью приступа и составляет от 10 до 50 мл (чаще 10-20 мл). Эффект должен наступить в конце вливания.
В связи с возможностью интоксикации (опасностью коллапса, фибрилляцией желудочков сердца) вводить препарат следует медленно.
Поскольку кальций выделяется из организма в течение 6-8 ч, инъекции целесообразно повторять 2-3 раза в сутки.
В межприступный период его препараты (глюконат, лактат, хлорид) применяют перорально в дозе 1-2 г в сутки после еды.
При кризе применяют также паратиреоидин - экстракт пара-щитовидных желез крупного рогатого скота в дозе 40-100 ед.
(2-5 мл) внутримышечно. Эффект наступает через 2-3 ч и длится сутки с максимумом действия через 18 ч. Паратиреоидин для поддерживающей терапии применяют ограниченно из-за возможности развития резистентности и возникновения аллергии. При необходимости проводят курсы лечения по 1,5-2 месяца с перерывами по 3-6 месяцев.
Важное значение в лечении имеют препараты витамина D, которые усиливают кишечную абсорбцию и реабсорбцию кальция в почечных канальцах, стимулируют его мобилизацию из костей. Наиболее эффективны препараты витамина D3, (IOН холекалыш-ферол, рксидевит, альфакальцидиол), которые выпускают в масляном растворе в дозах 1, 0,5 и ОД 5 мкг в капсулах для перорального применения, и l,25(OH)2D3-l,25(OH)2 (холекалышферол, рокалтрол), выпускаемый в тех же дозах и форме и в виде масляного раствора, содержащего 2 мкг/мл (0,1 мкг в 1 капле). В остром периоде суточная доза может составлять 2-4 мкг на 2 приема, поддерживающая - 0,5-1 мкг в сутки.
Сохраняет определенное значение лечение витамином D2 (эрго-калышферол) в спиртовом (200 тыс. МЕ/мл) и масляном (200, 50, 25 тыс. МЕ/мл) растворах.
В остром периоде назначают по 200-400 тыс. МЕ/мл, поддерживающая доза - 25-50 тыс. МЕ/мл.
Широко используется лечение 0,1%-ным масляным раствором дигидротахистерола (тахистином, AT-10 в капсулах), 1 мл которого содержит 1 мг дипшротахистерола. В остром периоде назначают по 1-2 мг каждые 6 ч, поддерживающая доза - 0,5-2 мл в день (подбирается индивидуально).
Лечение проводят под контролем исследования уровня кальция в крови во избежание передозировки и развития гиперкальциемии, которая сопровождается полиурией, сухостью во рту, жаждой, слабостью, головной болью, тошнотой, болями в животе, запорами.
При обнаружении гиперкальциемии необходимо отменить прием препаратов кальция и уменьшить дозу или отменить препараты, повышающие его содержание в крови, а также провести лечение, как при гиперкальциемическом кризе.
Для лечения гипопаратиреоза применяется диета, богатая солями кальция и магния (молоко, молочные продукты, овощи, фрукты) с ограничением фосфора (мясо). Отказ от мясных продуктов необходим особенно в период тетании. Желательно введение с пищей эргокалышферола, который содержится в рыбьем жире, сельди, печени, яичном желтке.
Для купирования гипомагнезиемии при пшопаратиреозе назначают сульфат магния в 25%-ном растворе по 10-20 мл в/м, при алкалозе - хлорид аммония до 3-7 г в сутки. Используют с целью симптоматической терапии седативные и противосудо-рожные препараты (хлоралгидрат в клизме, люминал, бромиды). При наличии ларингоспазма прибегают к интубации или трахеотомии.
Для создания «депо» кальция в организме применяют лиофили-зированный костный трансплантант, который пересаживают в мышцу спины, бедра или живота.
В последние годы практикуется трансплантация паращито-видных желез.
При псевдогипопаратиреозе лечение паратиреоидином неэффективно из-за нечувствительности к нему тканей-мишеней. Ги-покальциемию приходится у таких больных компенсировать введением препаратов кальция и витамина D. Обнадеживающие результаты дает применение активных препаратов витамина Д. При этом возможно возникновение гиперкальциемии при передозировке или индивидуальной повышенной чувствительности. В связи с редкостью псевдогипопаратиреоза и малым опытом лечения витамином D3 вопрос о его влиянии на метастатическую кальцификацию мягких тканей пока не ясен.
Диспансеризация.
Больные гипопаратиреозом должны находиться под регулярным диспансерным наблюдением эндокринолога. При налаженной стабильной терапии контроль уровня кальция и фосфора в крови необходим 1 раз в 4-6 месяцев. При первичном назначении терапии, смене лекарственных препаратов или подборе доз - контроль кальция и фосфора 1 раз в 7-10 дней.
Необходимо регулярное офтальмологическое наблюдение (катаракта); рентгенологическое исследование состояния черепа (кальцификация базальных ганглиев) и других костей по клиническим показаниям.
Трудоспособность. Трудоспособность зависит от тяжести процесса и степени медикаментозной компенсации. При латентной форме гипопаратиреоза и отсутствии явных тетаноидных приступов она частично сохранена (с определенными ограничениями). Рекомендуется работа, не связанная со значительными механическими, термическими и электрическими воздействиями на нервно-мышечный аппарат, противопоказан труд у движущихся механизмов, на транспорте. Необходимо исключение физического и нервно-психического перенапряжения. Нетрудоспособны больные с частыми тетаноидными приступами, а также при стойкой патологии со стороны центральной нервной системы и при нарушениях зрения из-за катаракты.
Псевдогипопаратирео з (греч. pseudē s ложный + гипопаратиреоз ; синоним: наследственная остеодистрофия Олбрайта, болезнь Олбрайта) - редкое наследственное заболевание костной системы, имитирующее гипопаратиреоз и характеризующееся нарушением обмена кальция и фосфора; часто сопровождается задержкой умственного и физического развития.
Что провоцирует / Причины Псевдогипопаратиреоза:
Причиной возникновения псевдогипопаратиреоза является врождённый дефект - нечувствительность периферических тканей к действию ПТГ.
Патогенез (что происходит?) во время Псевдогипопаратиреоза:
Полагают, что в основе псевдогипопаратиреоза лежит генетически обусловленная резистентность почек и скелета к действию паратгормона в результате дефекта комплекса специфический циторецептор - паратгормон - аденилатциклаза, что нарушает процесс образования в почках циклического 3", 5"-АМФ, являющегося внутриклеточным посредником действия паратгормона на метаболические процессы. Псевдогипопаратиреоз является генетически гетерогенным заболеванием. У части больных дефектен сам циторецептор, связывающий паратгормон (тип Ia псевдогипопаратиреоза), у других отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном бислое клеточной мембраны и функционально связывающего рецептор с аденилатциклазой (тип Iб псевдогипопаратиреоза). У некоторых больных наблюдается ферментативная недостаточность самой аденилатциклазы (псевдогипопаратиреоз II типа). Дефицит цАМФ, получающийся вследствие этих дефектов, ведет к нарушению синтеза специфических белков, определяющих биологический эффект паратгормона. Таким образом, теряется чувствительность органов-мишеней, в частности почек, к паратгормону. В результате уменьшается экскреция фосфора с мочой, возникает гиперфосфатемия, вторично развивается гипокальциемия. Так как при псевдогипопаратиреозе паращитовидные железы интактны, то в ответ на гипокальциемию, стимулирующую продукцию паратгормона, может развиться вторичный гиперпаратиреоз . Повышенное образование паратгормона не вызывает увеличения выведения фосфора и цАМФ с мочой из-за генетически обусловленной резистентности почечных канальцев к паратгормону, но сопровождается изменениями в костной ткани, характерными для гиперпаратиреоза, что свидетельствует о сохранении нормальной чувствительности остеокластов к паратгормону. При псевдогипопаратиреозе активность щелочной фосфатазы в сыворотке крови повышена или находится в пределах нормы (0,5-1,3 мкмоль неорганического фосфора на 1 мл сыворотки крови за 1 ч инкубации при 37°; определение по Боданскому). Все варианты псевдогипопаратиреоза представляют собой наследственное заболевание, характер наследования аутосомно-доминантный. Низкая плодовитость мужчин, страдающих псевдогипопаратиреозом, объясняет редкость его передачи от отца к сыну; женщины болеют в 2 раза чаще, чем мужчины.
Обычно при псевдогипопаратиреозе обнаруживают компенсаторную гиперплазию паращитовидных желез (наличие в них аденом не характерно). В костной ткани отмечают изменения, типичные для гиперпаратиреоза - диффузный остеопороз, появление кист (так называемые бурые опухоли, гигантоклеточные опухоли). Высвобождающийся из костей кальций откладывается в виде кальцинатов в подкожной клетчатке, а также в почках, мышцах, миокарде, стенках крупных артерий, конъюнктиве глаза и по периферии роговицы.
Симптомы Псевдогипопаратиреоза:
Клинические признаки псевдогипопаратиреоза сходны с симптомами идиопатического гипопаратиреоза . Отмечаются приступы тонических судорог, возникающие спонтанно или под влиянием каких-либо раздражителей. Кальцинаты в подкожной клетчатке проявляют тенденцию к изъязвлению. Подкожная оссификация часто выражена до такой степени, что имитирует оссифицирующий миозит. Характерны задержка умственного развития, отставание в росте, лунообразное лицо, ожирение и брахидактилия, особенно укорочение первой, четвертой и пятой пястных и плюсневых костей. Могут наблюдаться множественные экзостозы, дисхондроплазия, проявления вторичного гиперпаратиреоза в виде субпериостальной резорбции костей пальцев рук; изменения в эпифизах костей такие же, как при фиброзной остеодисплазии. Часто отмечают рвоту, а также гематурию вследствие образования оксалатных камней в мочевых путях, выявляют лентикулярную катаракту, гипоплазию зубной эмали.
У больных спсевдогипопаратиреозом наряду со снижением чувствительности к паратгормону органов-мишеней может наблюдаться резистентность к другим гормонам, зависимым от аденилатциклазной системы, например половых желез к гонадотропным гормонам, щитовидной железы к тиреотропному гормону, органов-мишеней к глюкагону и антидиуретическому гормону. Отмечается повышенная частота аутоиммунных болезней и диабета сахарного , наблюдаются гипотиреоз и гипертиреоз.
Выделяют также псевдопсевдогипопаратиреоз, который характеризуется отсутствием гипокальциемии, гиперфосфатемии, судорог и остеомаляции.
Диагностика Псевдогипопаратиреоза:
Диагноз в типичных случаях заболевания устанавливают у детей в 5-10 лет на основании характерной клинической картины, множественных аномалий развития костного скелета, наличия гипокальциемии, гиперфосфатемии, нормальной или повышенной активности щелочной фосфатазы в сыворотке крови, уменьшенного выделения кальция и фосфора с мочой, повышенного содержания паратгормона в крови. Наличие резистентности почечных канальцев к паратгормону подтверждает тест, основанный на определении количества фосфатов и цАМФ, выводимых с мочой. Отсутствие достоверного повышения содержания фосфатов и цАМФ в моче после введения больному паратгормона свидетельствует о резистентности почек к действию паратгормона. У больных с идиопатическим и послеоперационным гипопаратиреозом, наоборот, после внутривенного введения 200 ЕД паратгормона в моче содержание фосфатов и цАМФ в течение 4 ч увеличивается в 2-10 раз по сравнению с исходным уровнем. Выделение с мочой оксипролина у нелеченых больных с псевдогипопаратиреозом находится в норме или несколько повышено, а при гипопаратиреозе - понижено. Рентгенодиагностика псевдогипопаратиреоза основана на выявлении специфических изменений в костях и мягких тканях.
Псевдогипопаратиреоз в сочетании с гипогонадизмом у лиц женского пола необходимо дифференцировать с Шерешевского - Тернера синдромом , с которым псевдопсевдогипопаратиреоз сходен фенотипически. При синдроме Шерешевского - Тернера половой хроматин отсутствует, на месте яичников располагаются соединительнотканные тяжи, не определяемые при ректальном и ультразвуковом исследовании.
Лечение Псевдогипопаратиреоза:
Лечение при гипокальциемии заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови. Большое значение имеет терапия витамином D. Начальную дозу рассчитывают из 2000 МЕ/кг массы тела в сутки, но не более 100 000 МЕ в сутки. Во избежание передозировки препаратов витамина D необходим контроль за концентрацией кальция в крови каждые 3-7 дней в течение первых двух недель лечения и каждый месяц в течение последующих 2-3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2-3 месяца. Можно применять кальцитрин, дигидротахистерол, оксидевит, а также другие препараты активных форм витамина D. Диета с ограничением фосфора помогает нормализовать концентрацию кальция в крови и устранить симптомы вторичного гиперпаратиреоза. При недостаточности других желез внутренней секреции проводят заместительную терапию соответствующими гормонами. Лечение паратгормоном не эффективно. Для купирования судорожных приступов внутривенно вводят 10% раствор кальция хлорида или кальция глюконата; внутрь - 5-10% раствор кальция хлорида по 1 столовой ложке 3-4 раза в день: кальция глюконат, кальция лактат - до 10 г в день.
Прогноз при рациональной терапии благоприятный. Учитывая наследственный характер псевдогипопаратиреоза, необходимо медико-генетическое консультирование в отношении возможности появления псевдогипопаратиреоза у потомства.
Профилактика Псевдогипопаратиреоза:
К каким докторам следует обращаться если у Вас Псевдогипопаратиреоз:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Псевдогипопаратиреоза, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Euro lab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом . Клиника Euro lab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны . Посмотрите детальнее о всех услугах клиники на ее .
(+38 044) 206-20-00
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни . Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача , чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации , возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой . Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе . Также зарегистрируйтесь на медицинском портале Euro lab , чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни эндокринной системы, расстройства питания и нарушения обмена веществ:
| Аддисонический криз (острая недостаточность коры надпочечников) |
| Аденома молочной железы |
| Адипозогенитальная дистрофия (болезнь Перхкранца - Бабинского - Фрелиха) |
| Адреногенитальный синдром |
| Акромегалия |
| Алиментарный маразм (алиментарная дистрофия) |
| Алкалоз |
| Алкаптонурия |
| Амилоидоз (амилоидная дистрофия) |
| Амилоидоз желудка |
| Амилоидоз кишечника |
| Амилоидоз островков поджелудочной железы |
| Амилоидоз печени |
| Амилоидоз пищевода |
| Ацидоз |
| Белково-энергетическая недостаточность |
| Болезнь I-клеток (муколипидоз типа II) |
| Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия) |
| Болезнь Гоше (глюкоцереброзидный липидоз, глюкоцереброзидоз) |
| Болезнь Иценко-Кушинга |
| Болезнь Краббе (глобоидно-клеточная лейкодистрофия) |
| Болезнь Нимана - Пика (сфингомиелиноз) |
| Болезнь Фабри |
| Ганглиозидоз GM1 тип I |
| Ганглиозидоз GM1 тип II |
| Ганглиозидоз GM1 тип III |
| Ганглиозидоз GM2 |
| Ганглиозидоз GM2 тип I (амавротическая идиотия Тея - Сакса, болезнь Тея - Сакса) |
| Ганглиозидоз GM2 тип II (болезнь Сандхоффа, амавротическая идиотия Сандхоффа) |
| Ганглиозидоз GM2 ювенильный |
| Гигантизм |
| Гиперальдостеронизм |
| Гиперальдостеронизм вторичный |
| Гиперальдостеронизм первичный (синдром Конна) |
| Гипервитаминоз D |
| Гипервитаминоз А |
| Гипервитаминоз Е |
| Гиперволемия |
| Гипергликемическая (диабетическая) кома |
| Гиперкалиемия |
| Гиперкальциемия |
| Гиперлипопротеинемия I типа |
| Гиперлипопротеинемия II типа |
| Гиперлипопротеинемия III типа |
| Гиперлипопротеинемия IV типа |
| Гиперлипопротеинемия V типа |
| Гиперосмолярная кома |
| Гиперпаратиреоз вторичный |
| Гиперпаратиреоз первичный |
| Гиперплазия тимуса (вилочковой железы) |
| Гиперпролактинемия |
| Гиперфункция яичек |
| Гиперхолестеринемия |
| Гиповолемия |
| Гипогликемическая кома |
| Гипогонадизм |
| Гипогонадизм гиперпролактинемический |
| Гипогонадизм изолированный (идиопатический) |
| Гипогонадизм первичный врожденный (анорхизм) |
| Гипогонадизм первичный приобретенный |
| Гипокалиемия |
| Гипопаратиреоз |
| Гипопитуитаризм |
| Гипотиреоз |
| Гликогеноз 0 типа (агликогеноз) |
| Гликогеноз I типа (болезнь Гирке) |
| Гликогеноз II типа (болезнь Помпе) |
| Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз) |
| Гликогеноз IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) |
| Гликогеноз IX типа (болезнь Хага) |
| Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) |
| Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) |
| Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность) |
| Гликогеноз VIII типа (болезнь Томсона) |
| Гликогеноз XI типа |
| Гликогеноз Х типа |
| Дефицит (недостаточность) ванадия |
| Дефицит (недостаточность) магния |
| Дефицит (недостаточность) марганца |
| Дефицит (недостаточность) меди |
| Дефицит (недостаточность) молибдена |
| Дефицит (недостаточность) хрома |
| Дефицит железа |
| Дефицит кальция (алиментарная недостаточность кальция) |
| Дефицит цинка (алиментарная недостаточность цинка) |
| Диабетическая кетоацидотическая кома |
| Дисфункция яичников |
| Диффузный (эндемический) зоб |
| Задержка полового созревания |
| Избыток эстрогенов |
| Инволюция молочных желез |
| Карликовость (низкорослость) |
| Квашиоркор |
| Кистозная мастопатия |
| Ксантинурия |
| Лактацидемическая кома |
| Лейциноз (болезнь кленового сиропа) |
| Липидозы |
| Липогранулематоз Фарбера |
| Липодистрофия (жировая дистрофия) |
| Липодистрофия врожденная генерализованная (синдром Сейпа-Лоуренса) |
| Липодистрофия гипермускулярная |
| Липодистрофия постинъекционная |
| Липодистрофия прогрессирующая сегментарная |
| Липоматоз |
| Липоматоз болезненный |
| Метахроматическая лейкодистрофия |
| Микседематозная кома |
| Муковисцидоз (кистозный фиброз) |